nf-core/pathogensurveillance
Surveillance of pathogens using population genomics and sequencing
Introduction
nf-core/pathogensurveillance is a population genomics pipeline for pathogen identification, variant detection, and biosurveillance. The pipeline accepts paths to raw reads for one or more organisms and creates reports in the form of an interactive HTML document. Significant features include the ability to analyze unidentified eukaryotic and prokaryotic samples, creation of reports for multiple user-defined groupings of samples, automated discovery and downloading of reference assemblies from NCBI RefSeq, and rapid initial identification based on k-mer sketches followed by a more robust multi gene phylogeny and SNP-based phylogeny.
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. It uses Docker/Singularity/Conda to make installation trivial and results highly reproducible. The Nextflow DSL2 implementation of this pipeline uses one container per process which makes it much easier to maintain and update software dependencies. Where appropriate, these processes have been submitted to and installed from nf-core/modules in order to make them available to all nf-core pipelines, and to everyone within the Nextflow community!
On release, automated continuous integration tests run the pipeline on a full-sized dataset on the AWS cloud infrastructure. This ensures that the pipeline runs on AWS, has sensible resource allocation defaults set to run on real-world data sets, and permits the persistent storage of results to benchmark between pipeline releases and other analysis sources. The results obtained from the full-sized test can be viewed on the nf-core website.
Pipeline summary
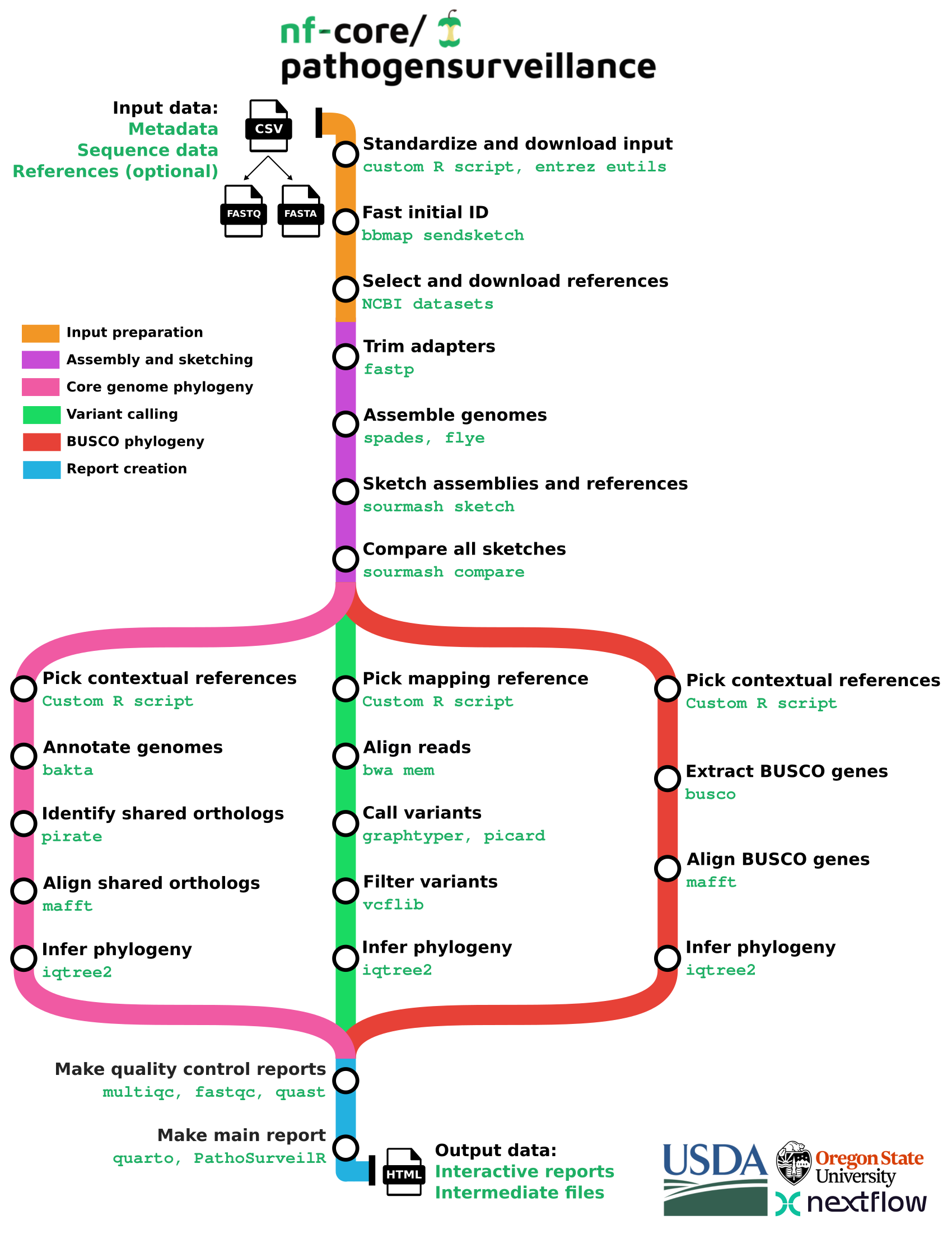
See for an example report generated by the pipeline.
Why use pathogensurveillance?
TL;DR: unknown gDNA FASTQ -> sample ID + phylogeny + publication-quality figures
Most genomic tools are designed to be used with a reference genome. Yet this is at odds with the work of pathogen diagnosticians, who often deal with unknown samples. Finding the right reference manually may be cumbersome and require a suprising amount of technical skill.
PathogenSurveillance picks a good reference genome for you. It does this through the program sourmash. In simple terms, this takes a sample’s DNA “fingerprint” and finds the closest match in a “DNA fingerprint library” spanning the tree of life. In more technical terms, the pipeline generates k-mer sketches from your reads assembled into genomes, then uses the identified reference to do a boilerplate, but robust phylogenetic analysis of your submitted samples.
In our experience, the pipeline usually chooses the best possible reference genome. At a minimum it will choose a reference that is good enough to build an informative phylogeny and allow you to see the contextual placement your samples.
Pathogensurveillance is designed to use as many types of genomic DNA input as possible. It works for common shortread and longread sequencing technologies and for both prokaryotes and eukaryotes. There is a good deal of emergent complexity required to work with such a broad sample range, but pathogensurveillance handles this automatically.
While PathogenSurveillance may be a useful tool for researchers of all levels, it was designed with those who may have limited bioinformatics training in mind. Pathogensurveillance is very simple to run. At a minimum, all that needs to be supplied is a .CSV file with a single column specifying the path to your sample’s sequencing reads. Other information is optional, but if provided will used to customize the output report or conditionally use particular reference genomes.
pathogensurveillance is particularly good for:
- unknown sample identification
- exploratory population analysis using minimal input parameters
- inexperienced bioinformatics users
- efficient parallelization of tasks
- repeated analysis (given caching) where you would like to add new samples to a past run
Note that pathogensurveillance works for non pathogens too!
pathogensurveillance is not designed for:
- viral sequence
- non gDNA datasets (DNA assembly fasta files, RNA-seq, RAD-seq, ChIP-seq, etc.)
- mixed/impure samples (this may change in future versions)
- Highly specialized population genetic analysis, or researchers who would like to extensively test parameters at each stage
Installation
The pipeline is automatically installed when run by nextflow, but you will need to install the following dependencies:
- nextflow
- At least one of the following container engines:
apptainer,charliecloud,docker,podman,sarus,shifter,singularity, orconda(only usecondaif no other option is available)
Quick start guide
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
Note that some form of configuration will be needed so that Nextflow knows how to fetch the required software.
This is usually done in the form of a config profile.
You can chain multiple config profiles in a comma-separated string.
In most cases you will include one profile that defines a tool to reproducibly install and use software needed by the pipeline.
This is typically one of docker, singularity, or conda.
Ideally conda should not be used unless docker or singularity cannot be used.
Profiles can also be used to store parameters for the pipeline, such as input data and pipeline options. Before using you own data, consider trying out a small example dataset included with the pipeline as a profile. Available test dataset profiles include:
test: Test profile of 1 small genome used to run the pipeline as fast as possible for testing purposes.test_serratia: Test profile of 10 serratia isolates from Williams et al. 2022 (https://doi.org/10.1038/s41467-022-32929-2)test_bordetella: Test profile of 5 Bordetella pertussis isolates sequenced with with Illumina and Nanopore from Wagner et al. 2023test_salmonella: Test profile of 5 salmonella isolates from Hawkey et al. 2024 (https://doi.org/10.1038/s41467-024-54418-4)test_boxwood_blight: Test profile of 5 samples of the boxwood blight fungus Cylindrocladium buxicola from LeBlanc et al. 2020 (https://doi.org/10.1094/PHYTO-06-20-0219-FI)test_mycobacteroides: Test profile of 5 Mycobacteroides abscessus samples from Bronson et al. 2021 (https://doi.org/10.1038/s41467-021-25484-9)test_bacteria: Test profile of 10 mixed bacteria from various sourcestest_klebsiella: Test profile of 10 K. pneumoniae and related species from Holt et al. 2015 (https://doi.org/10.1073/pnas.1501049112)test_small_genomes: Test profile consisting of 6 samples from species with small genomes from various sources.
Adding _full to the end of any of these profiles will run a larger (often much larger) version of these datasets.
For example, you can run the test_bacteria profile with the docker profile:
nextflow run nf-core/pathogensurveillance -r 1.1.0 -profile docker,test_bacteria -resume --outdir test_outputYou can see the samplesheets used in these profiles here:
https://github.com/nf-core/test-datasets/tree/pathogensurveillance
To run your own input data, prepare a samplesheet as described in the usage documentation section below and run the following command:
nextflow run nf-core/pathogensurveillance -r <REPLACE WITH VERSION> -profile <REPLACE WITH RUN TOOL> -resume --input <REPLACE WITH TSV/CSV> --outdir <REPLACE WITH OUTPUT PATH>Where:
<REPLACE WITH RUN TOOL>is one ofdocker,singularity, orconda<REPLACE WITH TSV/CSV>is the path to the input samplesheet<REPLACE WITH OUTPUT PATH>is the path to where to save the output<REPLACE WITH VERSION>is the version of the pipeline to run
Documentation
For more details and further functionality, please refer to the usage documentation and the parameter documentation. To see the results of an example test run with a full size dataset refer to the results tab on the nf-core website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
The following people contributed to the pipeline: Zachary S.L. Foster, Martha Sudermann, Camilo Parada-Rojas, Logan K. Blair, Fernanda I. Bocardo, Ricardo Alcalá-Briseño, Hung Phan, Nicholas C. Cauldron, Alexandra J. Weisberg, Jeff H. Chang, and Niklaus J. Grünwald.
Funding
This work was supported by grants from USDA ARS (2072-22000-045-000-D) to NJG, USDA NIFA (2021-67021-34433; 2023-67013-39918) to JHC and NJG, as well as USDAR ARS NPDRS and FNRI and USDA APHIS to NJG.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
For further information or help, don’t hesitate to get in touch on the Slack #pathogensurveillance channel (you can join with this invite).
Citations
Please cite use of nf-core/pathogensurveillance as follows:
Foster, ZSL, Sudermann, MA, Parada Rojas, CH, Blair, LK, Iruegas Bocardo, F, Dhakal, U, Weisberg, AJ, Phan, H, Chang, JH, Grunwald, NJ. PathogenSurveillance: an automated pipeline for population genomic analyses and pathogen identification. BioRxiv 2025 2025.10.31.685798. doi: https://doi.org/10.1101/2025.10.31.685798
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.